特許だけじゃない!新薬を守る再審査制度とデータ保護:知財戦略の「隠れた盾」を徹底解剖

はじめに:特許期間満了後の「第二の命」と市場独占の構造
株式会社IPリッチのライセンス担当です。日頃より、国内外の製薬企業やバイオベンチャー、そして投資家の皆様から、知的財産戦略やライセンス契約の評価(バリュエーション)に関するご相談を数多く承っております。その中で、特に創薬の現場において、特許権と同じくらい、あるいは局面によってはそれ以上に決定的な意味を持つ「もう一つの独占権」が存在することをご存じでしょうか。それが、本レポートの主題である「データ保護(Data Exclusivity)」および、日本独自の運用である「再審査制度」です。
一般的に、新薬開発には10年から15年の歳月と、数百億円から数千億円規模の莫大な投資が必要とされます 1。この巨額の先行投資を回収し、次なるイノベーションへの原資を確保するために、特許による市場独占は不可欠な要素です。しかし、特許権には「出願から20年」という厳格な期間制限が存在します。開発期間が長引けば長引くほど、あるいは薬事承認のプロセスが遅れれば遅れるほど、実際に製品を市場で独占販売できる期間(有効特許期間)は浸食されていきます。特許が切れた瞬間にジェネリック医薬品が参入し、売上が90%以上蒸発する「パテントクリフ(特許の崖)」は、製薬企業の経営を揺るがす最大のリスク要因です 2。
ここで、特許という「私権」とは異なるロジックで機能するのが、規制当局が行政処分として付与する「販売承認に基づく独占権」です。本レポートでは、特許権とは異なるこの「規制上の独占権(Regulatory Exclusivity)」に焦点を当て、日本、米国、欧州における制度の精緻な比較分析、バイオ医薬品時代におけるその重要性の高まり、そしてM&Aやライセンス交渉における事業価値評価への具体的なインパクトについて、網羅的かつ専門的な視点から徹底的に解説します。
知財の収益化と「PatentRevenue」が切り拓くキャリアの未来
本論に入る前に、現代の知財プロフェッショナルが直面しているキャリアの転換点について触れさせてください。昨今、企業の知的財産部門や研究開発部門では、単に特許を出願し権利を守るという従来の「守り」の機能だけでなく、保有する特許群や技術ノウハウを積極的にライセンスアウトし、あるいは非中核資産を売却することで新たなキャッシュフローを生み出す「知財の収益化」が、経営上の最重要課題として浮上しています。
こうした知財活用の高度化に伴い、ライセンス交渉やIPバリュエーションをリードできる専門人材への需要は、かつてないほど高まっています。もしあなたが、これまでに培った知財スキルや契約実務の経験を、よりダイナミックな「収益化」や「事業開発」のフィールドで試したいとお考えなら、知財専門のハイクラス求人プラットフォーム「PatentRevenue」への登録を強くお勧めします。ここには、一般の転職サイトには出回らない非公開のライセンス担当ポジションや、IP戦略コンサルタントの求人が多数掲載されており、知財のプロフェッショナルとしてのキャリアを飛躍させる機会に溢れています。
(求人登録はこちら:https://patentrevenue.com)
第1章:パテントクリフの脅威と「データ保護」という防波堤
市場独占の崩壊とジェネリック参入のメカニズム
医薬品ビジネスにおいて、一つの製品が稼ぎ出す収益の曲線は、特許満了日を境に劇的な変化を遂げます。いわゆる「ブロックバスター」と呼ばれる年間売上10億ドル超の大型新薬であっても、特許保護が失われ、ジェネリック医薬品(後発薬)の参入を許せば、その市場シェアは急速に侵食されます。米国市場のデータによれば、最初のジェネリック参入から数ヶ月以内に、先発薬の処方量は激減し、価格競争によって市場規模そのものが縮小する現象が常態化しています 3。
しかし、ここで重要なのは「特許が切れた瞬間に、無条件でジェネリックが参入できるわけではない」という事実です。ジェネリック医薬品が市場に出るためには、規制当局(日本のPMDA、米国のFDA、欧州のEMAなど)から製造販売承認を得る必要があります。通常、医薬品の承認を得るには、安全性と有効性を証明するための膨大な臨床試験データ(治験データ)が必須となります。先発メーカーはこのデータの構築に長い年月と巨額の費用を投じてきましたが、ジェネリックメーカーが同じ規模の臨床試験を自前で行っていては、安価な医薬品を提供することは不可能です。
そこで、多くの国の規制制度では、ジェネリックメーカーが承認申請を行う際、先発メーカーが蓄積した臨床試験データを「参照」あるいは「引用」することを認めています。これにより、ジェネリックメーカーは試験を省略し、開発コストを抑えることができるのです。しかし、この「参照」を無制限に認めてしまえば、先発メーカーのデータ構築への投資インセンティブが損なわれます。ここで機能するのが「データ保護(Data Exclusivity)」です。これは、規制当局が先発薬の臨床試験データを「他社が参照することを禁じる」、あるいは「参照して申請することを一定期間受け付けない」という行政的な保護期間を指します 1。
特許とデータ保護の法的な乖離と補完性
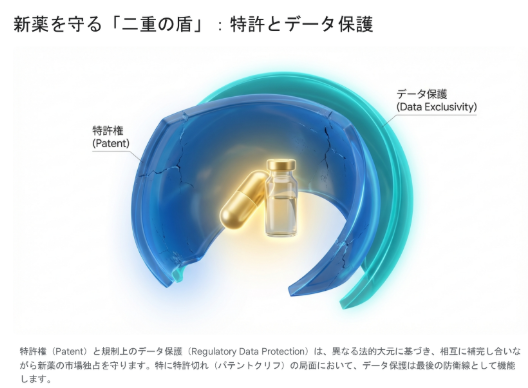
データ保護期間中は、たとえ当該医薬品の物質特許が無効になったり、期間満了を迎えたりしていたとしても、ジェネリックメーカーは承認申請に必要なデータを提出できない(参照できない)ため、事実上の市場参入が不可能となります。つまり、データ保護は特許権とは独立した「第二の独占権」として機能するのです。両者の違いを理解することは、知財戦略の根幹に関わります。
まず、根拠法の違いがあります。特許権は特許法に基づき、発明(物質、製法、用途など)を保護対象として特許庁が出願日を起点に付与します。一方、データ保護は薬機法(日本)やFD&C法(米国)などの規制法に基づき、臨床試験データ(安全性・有効性の証明パッケージ)を対象として規制当局が承認日を起点に付与します。
次に、期間の起算点の違いが決定的です。特許は「出願日」から20年であるため、開発期間が長引くほど市場での独占期間(有効特許期間)は短くなります。これに対し、データ保護は「承認日(販売開始日)」からカウントダウンが始まるため、開発が難航し特許期間が残り少なくなった医薬品にとっては、市場投入後の一定期間を確実に守る強力な命綱となります 5。
さらに、安定性と確実性の違いも見逃せません。特許権は、競合他社からの無効審判(Invalidation Challenge)や訴訟によって、事後的に無効化されるリスクを常に抱えています。特に米国では、パラグラフIV認証を通じたジェネリックメーカーからの特許挑戦が日常的に行われています。しかし、データ保護は規制当局が法律に基づき自動的かつ行政的に付与する期間であるため、訴訟によって覆される可能性が極めて低く(制度自体の違憲訴訟などを除き)、ビジネス上の予見可能性が非常に高いという特徴があります 6。投資家やライセンスの買い手が、特許の残存期間以上にデータ保護期間を重視するケースがあるのは、この「確実性(Certainty)」に価値があるからです。
第2章:日本における「再審査制度」の深層
安全性監視から生まれた「事実上の独占」
日本には、欧米のような「データ保護制度(Data Exclusivity)」という名称の明示的な条項は法律上存在しません。しかし、医薬品医療機器等法(薬機法)第14条の4に基づく**「再審査制度」**が、実質的にデータ保護と同じ、あるいはそれ以上の機能を果たしています 5。
この制度の本来の趣旨は、市場独占の保護ではなく「安全性確保」にあります。新薬は治験という限られた条件下でのデータに基づいて承認されますが、市販後に不特定多数の患者に使用された場合の安全性や有効性を再確認する必要があります。そのため、承認後の一定期間(再審査期間)は、製造販売業者が使用成績調査(PMS)などを実施し、その結果を報告することが義務付けられています。
この「再審査期間中」は、他社(ジェネリックメーカー)が同一成分の医薬品を承認申請しようとしても、再審査に必要なデータと同等のデータ(つまり大規模な臨床試験データ)を自前で提出しない限り、承認されません。ジェネリックメーカーにとって自前での臨床試験実施は経済的に合理的ではないため、結果として再審査期間が終了するまでジェネリックは参入できず、先発メーカーの市場独占が維持される仕組みとなっています 5。
期間設定の精緻なカテゴリー
再審査期間は一律ではなく、その医薬品の新規性や医療上の必要性に応じて細かく設定されています 6。
- 希少疾病用医薬品(オーファンドラッグ):10年
- 患者数が5万人未満などの要件を満たし、希少疾病用医薬品の指定を受けた場合、最長である10年間の再審査期間が付与されます。開発回収リスクが高いニッチな領域への参入を促すための最強のインセンティブであり、特許期間が切れていても10年間は独占が約束されます。
- 新有効成分含有医薬品(NCE):8年
- 日本で初めて承認される新しい有効成分を含む医薬品には、原則として8年間の期間が設定されます。これは世界の主要国と比較しても標準的かつ十分な期間と言えます。
- 新配合処方・新投与経路:6年(または4年)
- 既存の成分であっても、新たな配合や投与経路(例:注射剤から経口剤へ)を開発した場合、その医療上の有用性に応じて6年の期間が付与されます。ただし、医療上の有用性が新有効成分ほど高くないと判断される場合などは4年となることもあります。
- 新効能・新用量:4年〜
- 既に承認されている薬に新しい適応症(効能)を追加した場合、その部分について4年間の調査期間が課されます。これにより、適応拡大に対するインセンティブが働きます。
- 小児用法の追加に伴う延長
- 特定の条件下で小児向けの用法・用量を追加する開発を行う場合、再審査期間が延長されるケースもあります。
このように、日本の再審査制度は「安全性監視」という建付けを取りながらも、実質的には開発の難易度や貢献度に応じた「イノベーションへの報酬」として機能しており、知財戦略上、特許と並ぶ車の両輪として扱われています。
第3章:グローバル・ランドスケープ — 米国Hatch-Waxman法と欧州の精緻なルール
日本の再審査制度をより深く、相対的に理解するためには、世界最大の医薬品市場である米国と、高度に統合された欧州市場の制度との比較が不可欠です。各地域の制度設計には、イノベーションの促進と医療費抑制という、相反する政策目的のバランスをどう取るかという思想の違いが色濃く反映されています。
米国:Hatch-Waxman法による競争と保護の均衡
米国では、1984年に制定されたHatch-Waxman法(ハッチ・ワックスマン法、正式名称:Drug Price Competition and Patent Term Restoration Act)が、現代の医薬品知的財産システムの基礎を築きました。この法律は、先発メーカーとジェネリックメーカーの間の「大いなる妥協(The Great Compromise)」として知られています 9。
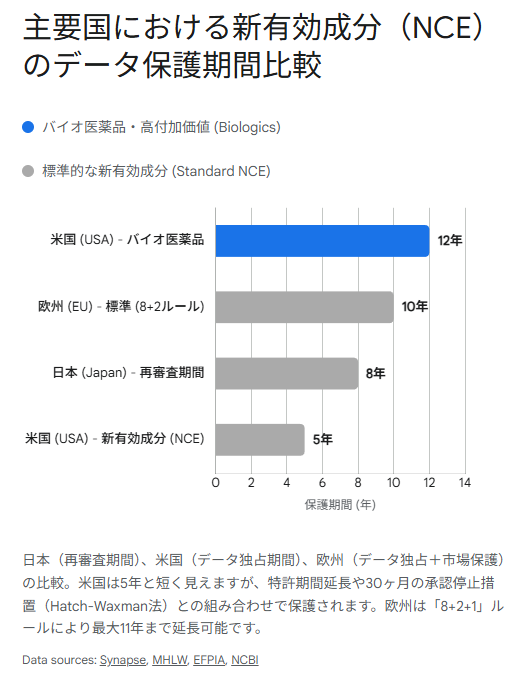
米国におけるデータ保護(Exclusivity)の期間は、一見すると日本や欧州よりも短く設定されています。
- 新有効成分(NCE):5年8
- 全く新しい成分の薬については、FDA承認から5年間、データ保護が付与されます。この期間中、FDAはジェネリック薬の簡略承認申請(ANDA)を受け付けることができません(ただし、特許挑戦を伴うパラグラフIV認証を行う場合は4年経過後から申請可能)。
- 新規臨床試験(新しい適応、剤形など):3年
- 既存薬の改良(SNDAs)に対しては、3年間の保護が付与されます 11。
「たった5年か」と思われるかもしれませんが、米国システムの特徴は、データ保護単体ではなく、特許制度との強力な連携にあります。Hatch-Waxman法は、特許期間の延長(Patent Term Extension)を認めると同時に、ジェネリックメーカーが特許に挑戦する際の手続きを整備しました。特筆すべきは、先発メーカーがジェネリック申請に対して特許侵害訴訟を提起した場合、FDAによるジェネリック承認が最大**30ヶ月間自動的に停止される(30-month stay)**という規定です 11。この「30ヶ月ステイ」と「5年のデータ保護」が組み合わさることで、実質的な独占期間は長期化し、先発メーカーの利益が守られる構造になっています。
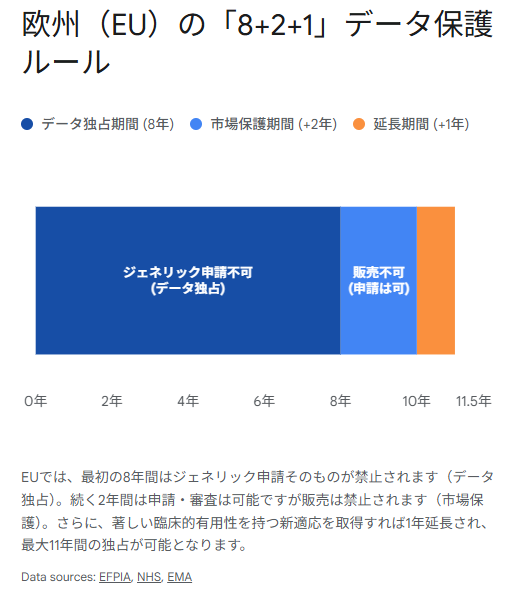
欧州:「8+2+1」ルールの重層的保護
一方、欧州連合(EU)は、**「8+2+1」**と呼ばれる独特かつ合理的なシステムを採用しています 4。このルールは、データ独占と市場保護を明確に区分し、さらにイノベーションへのボーナスを用意することで、企業の開発意欲を刺激する設計となっています。
- データ独占期間(Data Exclusivity):8年
- 承認から8年間は、ジェネリックメーカーは先発薬のデータを参照して販売承認申請(MAA)を行うこと自体が禁じられています。つまり、申請書類を提出することすらできません 14。
- 市場保護期間(Market Protection):+2年
- 8年が経過するとジェネリックメーカーは申請が可能になり、審査も進めることができます。しかし、承認が下りたとしても、さらに2年間は製品を市場で販売(上市)することができません。これにより、合計10年間の市場独占が事実上保証されます 4。
- 適応拡大による延長:+1年
- 最初の8年以内に、既存の治療法と比較して「著しい臨床的有用性(Significant Clinical Benefit)」を持つ新しい適応症(New Therapeutic Indication)を取得した場合、市場保護期間がさらに1年延長され、最大11年の保護が得られます 1。
この「著しい臨床的有用性」の要件は厳格ですが、成功すればブロックバスター薬の寿命をさらに延ばすことができるため、欧州でのライフサイクルマネジメント(LCM)戦略において極めて重要なマイルストーンとなります 14。単純な適応追加では認められず、安全性の大幅な向上や、これまで治療法がなかった疾患への効果など、明確な科学的根拠が求められます。
アジア諸国との比較:韓国の独自性
主要国以外の動向にも目を向けると、韓国におけるデータ保護制度の運用も興味深い示唆を含んでいます。調査データによると、韓国では新薬の承認から後発薬の参入までの「指定ギャップ(Designation Gap)」が他国と比較して顕著に長い(中央値で約59ヶ月)という分析結果があります 16。これは制度的なデータ保護期間だけでなく、薬価収載プロセスや特許係争の独自性が絡み合った結果と考えられますが、アジア市場への展開を考える際には、単なる「法定期間」だけでなく、こうした「実質的な参入障壁(Lag Time)」を考慮に入れる必要があります。
第4章:バイオ医薬品時代と「12年」の攻防
製薬業界の技術トレンドは、低分子医薬品から、抗体医薬や遺伝子治療などのバイオ医薬品へと急速にシフトしています。このパラダイムシフトに伴い、データ保護の重要性はかつてないほど高まっています。その象徴が、米国の**バイオ医薬品価格競争・イノベーション法(BPCIA)で定められた「12年」**という長期間のデータ保護です 1。
なぜバイオ医薬品は「12年」なのか?
低分子薬の5年に対し、なぜバイオ医薬品には12年もの保護が必要なのでしょうか。その理由は、バイオ医薬品の開発・製造の特殊性にあります。
バイオ医薬品は、生きた細胞を用いて製造されるため、その構造は極めて複雑かつ不均一です。「プロセスがプロダクトである」と言われるように、製造工程のわずかな違いが最終製品の品質に影響を与えます。そのため、開発リスクが高く、製造設備の投資額も巨額になります。
研究によれば、バイオ医薬品のポートフォリオが損益分岐点を超えて投資を回収するには、平均して12.9年から16.2年かかるとの試算があります 19。もし保護期間が低分子薬と同じ5年であれば、投資回収が完了する前にバイオシミラー(バイオ後続品)が参入してしまい、イノベーションへの再投資サイクルが断絶する恐れがありました。
また、特許による保護の限界も背景にあります。低分子薬は化学構造が明確であるため、物質特許で強力に守ることができます。しかし、巨大な分子であるバイオ医薬品は、アミノ酸配列が同じでも立体構造や糖鎖修飾が異なる場合があり、特許の権利範囲を厳密に定義し、侵害を立証することが技術的に困難な場合があります。また、競合他社が特許を回避する「デザインアラウンド」を行いやすいという側面もあります。そのため、物質そのもののデータ保護期間を法的に長く設定することで、特許の不確実性を補完し、開発インセンティブを維持するという政策判断がなされたのです 20。
ケーススタディ:ヒュミラとエンブレルの要塞
この「12年」のデータ保護と特許戦略を組み合わせることで、巨額の収益を守り抜いた事例として、アッヴィ社の「ヒュミラ(Humira)」とアムジェン社の「エンブレル(Enbrel)」が挙げられます。
世界で最も売れた薬の一つであるヒュミラは、物質特許の満了が迫る中で、製法特許や投与方法に関する特許を多数出願し、いわゆる「パテント・シケット(特許の茂み)」を形成しました。これに加え、データ保護期間を最大限に活用することで、米国でのバイオシミラー参入を2023年まで遅らせることに成功しました 21。その結果、特許切れが懸念された時期を超えてもなお、年間200億ドル規模の売上を維持し続けました。
エンブレルの事例も劇的です。当初の特許は2012年頃に切れる予定でしたが、アムジェン社は新たな特許を取得し、さらにデータ保護のロジックを組み合わせることで、バイオシミラーの参入を2029年まで阻止する法的地位を確立しました 23。裁判所はアムジェン社の主張を認め、競合であるサンド社のバイオシミラー販売を差し止めました 25。これにより、エンブレルは発売から30年近くにわたり独占的な地位を享受することになります。これは、知財戦略と規制戦略の融合がいかに強力な「参入障壁」を築き得るかを如実に示しています。
第5章:M&Aとライセンスにおける「データ保護」の価値評価(Valuation)
最後に、IPリッチのライセンス担当として最も強調したいのが、**「Valuation(事業価値評価)」**におけるデータ保護の役割です。M&Aやライセンス契約のデューデリジェンス(資産査定)において、これまでは「特許の残存期間」が価値算定の主役でした。しかし、現代の高度なバリュエーション実務においては、特許権のリスクとデータ保護の確実性を天秤にかけた、より精緻なモデルが求められています 27。
「不確実な20年」より「確実な8年」
投資家や買収側企業(買い手)は、将来のキャッシュフローの「確実性(Certainty)」に対してプレミアムを支払います。特許は20年の期間がありますが、常に無効審判のリスクに晒されています。特に、高額な医薬品ほどジェネリックメーカーからの特許挑戦を受ける確率は高まります。ある日突然、裁判所の判決で特許が無効となり、キャッシュフローがゼロになるリスクを常に割引率(Discount Rate)に織り込む必要があります。
対照的に、データ保護期間(再審査期間)は、規制当局が付与する行政処分であるため、第三者が裁判で覆すことは極めて困難です。つまり、データ保護期間中のキャッシュフローは、特許期間中のそれよりも「リスクが低い(確実性が高い)」と評価されます。
例えば、特許があと3年で切れるが、希少疾病用医薬品としての再審査期間があと7年残っている新薬候補があるとします。従来の特許中心の評価では「残り3年の価値」とみなされがちですが、実務的には「残り3年の特許独占+その後の4年間のデータ保護独占=計7年の高確度キャッシュフロー」として評価されます。この「追加の4年間」が生み出す価値(NPV)は、数百億円規模に達することもあり、ディールの成否やロイヤリティ料率を決定づける要因となります 3。
資産価値の底上げ効果
また、特許戦略が失敗した場合の「保険」としての価値も見逃せません。開発中の新薬において、物質特許の成立が危ぶまれるケースや、先行技術が存在し特許の有効性に疑義があるケースでも、もしその薬がオーファンドラッグ指定やNCEとしての要件を満たしていれば、最低限の独占期間(日本なら8年または10年、米国なら7年または5年)は確保できます。この「フロア(最低保証)」があることで、リスクの高い初期段階のライセンス契約でも、一定の一時金(アップフロント)やマイルストン支払いが正当化されるのです。
さらに、データ保護制度が整備されつつある新興国市場における価値も見逃せません。知的財産権の行使(エンフォースメント)が不安定な国であっても、薬事当局によるデータ保護が機能していれば、現地企業によるコピー製品の参入を水際で防ぐことができます 29。これはグローバルライセンス契約において、対象地域の範囲(Territory)を広げ、契約総額を最大化するための重要な交渉材料となります。
結論:複合的な独占戦略の構築に向けて
本レポートで見てきたように、新薬を守る戦いは、もはや「特許出願」だけでは完結しません。「特許権」という鋭いが折れることもある剣と、「データ保護(再審査制度)」という範囲は限定的だが極めて堅牢な盾。この2つの武器を、各国の制度差(米国の12年、欧州の+1年、日本の再審査)というフィールドの特性に合わせて巧みに組み合わせることで初めて、製品のライフサイクル価値(Lifetime Value)を最大化することが可能になります。
特に、特許期間が短くなりがちな開発期間の長い革新的新薬や、特許による保護が難しいバイオ医薬品においては、開発の初期段階から「どのような適応症で承認を取るか(オーファン指定を狙うか)」、「どのタイミングで小児データを出すか(延長を狙うか)」といった規制戦略(Regulatory Strategy)を知財戦略と一体化させて策定することが求められます。
私たちIPリッチは、単なる特許管理の枠を超えて、こうした規制制度や事業価値評価まで含めた包括的な知財収益化戦略を支援しています。お手持ちの技術や医薬品候補物質が持つ「隠れた価値」を再発見し、最強の独占戦略を構築するために、ぜひ私たちの知見をご活用ください。
参考文献
- 17 Synapse. “Which countries offer the longest data exclusivity for biologics?”
- 16 TandFOnline. “Drug data exclusivity Japan vs US vs EU comparison”
- 11 Fasken. “United States Exclusivities for Innovators”
- 5 Nishimura & Asahi. “Pharmaceutical Intellectual Property and Competition | Japan”
- 29 Katten. “Beyond borders 2025: The top five considerations…”
- 27 Drug Patent Watch. “The Patent Playbook: A Strategic Guide to M&A”
- 8 厚生労働省 (MHLW). “米国・欧州・日本・中国・韓国のデータ保護期間比較”
- 1 Scendea. “Data exclusivity vs patent protection pharma valuation”
- 3 EY. “Navigating Pharma Loss of Exclusivity”
- 4 EFPIA. “Regulatory Data Protection (RDP)”
- 12 SPS NHS. “Understanding data exclusivity and market protection”
- 13 EMA. “Presentation: Data exclusivity, market protection, orphan and paediatric rewards”
- 9 Fish & Richardson. “Hatch-Waxman 101”
- 2 Accessible Meds. “The Hatch-Waxman 180-Day Exclusivity Incentive”
- 13 EMA. “Data exclusivity and Market Protection”
- 1 Scendea. “US biologics data exclusivity 12 years rationale”
- 19 Nature Biotechnology (via Duke). “The market exclusivity period required for a pioneer biologic”
- 20 I-MAK. “Biologics Biosimilars Guide”
- 18 CRS Report. “Intellectual Property Rights and the Pharmaceutical Industry”
- 3 EY. “Navigating Pharma Loss of Exclusivity”
- 28 Bluestar BioAdvisors. “Valuation Case Studies”
- 1 Scendea. “Pharmaceutical research and development costs and protections”
- 15 European Commission. “Guidance on elements required to support the significant clinical benefit”
- 3 EY. “Navigating Pharma Loss of Exclusivity”
- 6 NCBI. “Data Exclusivity in Japan: Re-examination Period”
- 21 Nasdaq. “Has AbbVie Successfully Navigated Top-Line Growth Post-Humira LOE”
- 22 NCBI. “Humira data exclusivity impact revenue”
- 23 I-MAK. “Enbrel Report”
- 25 Pharmaceutical Technology. “Sandoz challenges Amgen’s Enbrel patents”
- 24 GaBI Online. “Etanercept biosimilars delayed until 2029 in US”
- 26 Center for Biosimilars. “NJ Court Decision Means 3 Decades of Product Exclusivity for Enbrel”
- 1 Scendea. “EU data exclusivity significant clinical benefit example”
- 6 NCBI. “Data Exclusivity in Japan: Re-examination Period”
- 7 NPRA. “Japan regulatory system for pharmaceutical products”
※本記事はAIによって作成されました。情報の正確性には万全を期していますが、法的な助言を構成するものではありません。個別の案件については専門家にご相談ください。
関連記事
-
 秘密保持契約(NDA)と営業秘密の違い:知財収益化を守るための完全ガイド
秘密保持契約(NDA)と営業秘密の違い:知財収益化を守るための完全ガイド -
ドメイン名の落とし穴:サイバースクワッティングとタイプスクワッティングの脅威と実務的対策
-
特許にならないアイデア:何が「発明」にならないのか? 特許要件と収益化の完全ガイド
-
YKKの「ブラックボックス戦略」と知財経営:1200件超の特許が支える世界シェアトップの秘密
-
商標権と著作権の境界線:オセロとリバーシから学ぶ知財戦略の攻防
-
サーバーの向こう側へ:ドワンゴ対FC2・最高裁判決が拓いた「国境なき知財」の新時代
-
眠れる巨人を揺り起こせ:知財収益化の深層と、IBM・FRONTEOが描いた再生の軌跡
-
パロディ表現と著作権法の境界線:ビジネスにおける法的リスクと知財戦略の全貌